




近期, 巴西通过世贸组织网站发布第657号RDC决议 (RESOLUÇÃO DE DIRETORIA COLEGIADA),该决议规定了医疗器械软件 (Software as a Medical Device,简称SaMD) 监管相关的事项要求,并将于2022 年7月1日起正式生效。
主要内容简介如下:
1、新规涵盖的医疗器械软件范围
医疗器械软件(Software as a Medical Device - SaMD) 在RDC657号决议中给出了定义。医疗器械软件属于医疗器械范畴,具有一个或多个医疗用途,并且无需成为医疗器械硬件的组件即可完成预期用途。
需注意的是以下5种情况不属于RDC 657法规的监管范畴:
(一)健康软件,旨在鼓励和保持健康的软件,如体育锻炼等健康活动,但不能用于预防、诊断、治疗、康复或避孕;
(二)列在国家卫生监督局 (Anvisa) 未受管制产品清单中的软件;
(三)专用于健康服务领域的行政和财务管理的软件;
(四)处理人口统计和流行病学医学数据,无临床诊断或治疗目的的软件;
(五)运行在接受卫生监督的医疗器械上的软件。
2、监管总则
RDC 657号决议中列出了4大监管总则,概要如下:
(一)医疗器械专用配件的医疗应用软件和具有医疗应用的嵌入式软件应该与医疗器械一起接受相关卫生监督制度的监管。
(二)医疗器械软件 (SaMD) 必须符合RDC 185号决议或后续相关法规。
(三)由卫生部门内部(in house)开发的并仅供卫生部门、总部或分支机构使用的医疗器械软件 (SaMD),在属于风险等级I和II的情况下,只要它们不干扰可受合规化处理的医疗器械的功能,则不需通过巴西卫生监督局合规化。但是需要注意如下事项:
- 禁止在未向巴西卫生监督局正式注册的情况下将内部开发的医疗器械软件商业化或捐赠。
- 卫生部门应该持有医疗器械软件内部开发确认的完整记录,包括证明其内部开发和更新历史的文件。
- 如果卫生部门内部开发的医疗器械软件没有内部开发确认的至少10年内的完整记录,应视为未注册,并受到相应的卫生行政处罚。
- 确认证据必须足以保证准确性、可靠性和预期性能,能够辨别出无效记录或更改记录的能力。
- 卫生部门将在本决议 (RDC 657) 发布后的2年内实现对医疗器械软件内部开发的确认。
-
每个菜单项和指令的含义在使用说明中用葡萄牙语解释; -
不提供给非专业人士或在家庭环境中使用; -
企业的风险管理认为这是可接受的风险; -
在使用说明中写明了语言能力是操作员的先决条件之一。
3、风险等级为I和II类医疗器械软件的备案要求
(一) 医疗器械软件合规化必须符合医疗器械的一般规定,特别是2001年10月22日RDC 185号决议和2015年8月26日 RDC 40号决议,包括其后续修订。
(二)对于风险等级为I和 II 的医疗器械软件,必须提交软件备案申请表,该申请表可在巴西卫生监督局的电子门户网站上获取。
(三)风险等级为 I 和 II 的医疗器械软件,技术档案中必须包含如下事项:
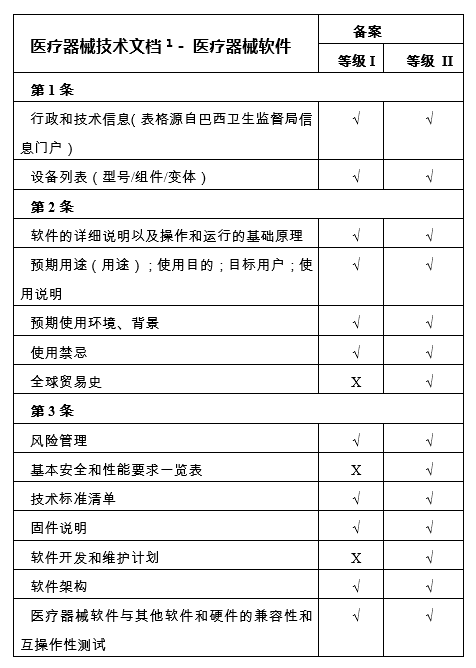
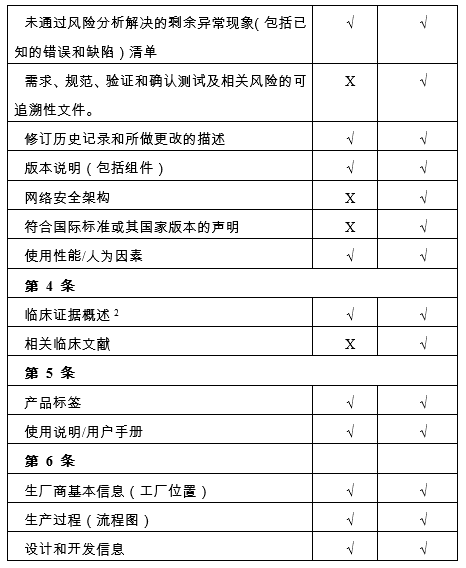
备注:
1、医疗器械技术文档结构是符合国际医疗器械监管机构论坛-IMDRF/RPS WG/N9(第3版)最新版本:2019-非体外诊断设备市场授权目录(nlVD MA ToC)发布的文件,并会根据后续版本进行调整。
2、仅适用于当证明安全性和性能、技术创新和新的使用适应症而需要临床证据的情况,根据巴西现行的临床试验卫生法规,应提交一份具体声明。
4、风险等级为III 和 IV类 医疗器械软件的注册要求
医疗器械软件风险等级 III 和 IV 在注册过程中提交的技术报告中,除符合I和II类医疗器械软件备案要求外,还必须包括:
(一) 软件架构;
(二) 硬件架构以及最低的和建议的技术要求;
(三) 系统平台;
(四) 与其他医疗产品包括其他软件或体外诊断产品的兼容性、互操作性和互通性,
(五) 网络安全架构信息和控制;
(六) 验证和确认;
(七) 风险管理;
(八) 发现的剩余缺陷以及解决的方法;
(九) 临床评价和临床有效性,包括用于处理生成预防、诊断、治疗、生理监测、康复或避孕的建议及其临床或科学依据的算法和 (或) 程序的通用描述;
(十) 符合国际标准或其国家标准的声明,声明必须至少包括以下版本或其更新版本:
- IEC 62304:2006 - 医疗器械软件——软件生命周期过程;
- IEC 62366-1:2015医疗器械——第 1 部分:可用性工程在医疗器械中的应用;
- ISO 14971:2007 医疗器械——风险管理在医疗器械中的应用。
如果递交的声明没有引用以上的任何标准,则需提供产品生命周期的描述、医疗器械软件的可用性研究报告 (人为因素) 风险管理报告技术证明文件,证明与所缺标准相对应的产品的安全性和有效性。如果该医疗器械软件有特定的国际或国家技术标准,其测试和验证报告可用于证明产品的安全性和有效性,但仍以巴西卫生监督局的技术分析为通过条件。
5、医疗器械软件若出现以下情况需进行变更注册
医疗器械软件的更新必须符合2020年3月6日RDC 340号决议中的相关规定,包括其后续修订内容。
下列情况,需申请变更注册:
(一) 具有新的临床功能或适应症的更新;
(二) 显著影响到医疗器械临床功能、临床安全性、疗效、性能的更新;
(三) 视觉识别的错误描述,使软件不再能被巴西卫生监督局从图像中识别出来。
为简单维护而进行的更新,不会受到巴西卫生监督局的约束,例如不改变视觉标识的视觉更改、错误更正、程序修改,或者不影响使用指征的信息安全、医疗器械软件的有效性或患者安全等其他方面的更改。
6、法规生效日期
RDC 657号决议将于2022 年 7 月 1 日起正式生效。